최근 물리학 리뷰 리서치에 실린 기사에서 우리는 딥러닝이 실제 시스템의 양자역학의 기본 방정식을 해결하는 데 얼마나 도움이 되는지 보여 주었다. 이는 과학적으로 중요한 질문일 뿐만 아니라 향후 실용화 될 수 있기 때문에 연구자들이 실리코(silico)에서 새로운 물질과 화학 합성물을 실험실에서 만들기 전에 시제품으로 만들 수 있도록 한다. 오늘 우리는 또한 이 연구의 코드를 발표하여 계산 물리학과 화학 커뮤니티가 우리의 작업을 기반으로 광범위한 문제에 적용할 수 있기를 희망한다. 우리는 새로운 신경망 구조인 페르미온 신경망(Fermionic Neural Network) 또는 페르미네트(FermiNet)를 개발하였다. 이 구조는 화학 결합의 기본 구성 요소인 대규모 전자 집합의 양자 상태를 모델링하는 데 적합하다. 페르미넷은 유용할 만큼 정확한 제1원리에서 원자와 분자의 에너지를 계산하기 위한 딥러닝의 첫 시연이었으며, 현재까지 가장 정확한 신경망 방법으로 남아 있다. DeepMind의 AI 연구에서 개발된 도구와 아이디어가 자연과학의 근본적인 문제를 해결하는 데 도움이 되기를 바라며, FermiNet은 단백질 접힘(protein folding), 유리 역학(glassy dynamics), 격자 양자 색역학(lattice quantum chromodynamics) 및 기타 여러 프로젝트에 대한 우리의 연구에 동참하여 이러한 비전을 실현하기를 바란다.
양자역학의 간략한 역사
"양자역학"을 언급하면 여러분은 다른 어떤 것보다 더 혼란을 불러일으킬 수 있다. 이 용어는 역설적으로 살아있는 것과 죽은 것 모두일 수 있는 슈뢰딩거의 고양이와 어떻게든 파동인 근본적인 입자들의 이미지를 떠올리게 한다. 양자 시스템에서 전자와 같은 입자는 고전적인 설명처럼 정확한 위치를 가지고 있지 않다. 대신, 그 위치는 확률 구름에 의해 설명할 수 있다. 구름은 허용 가능한 모든 곳에 얼룩져 있습니다. 이러한 반직관적인 상태는 리차드 파인먼으로 하여금 "당신이 양자역학을 이해하고 있다고 생각한다면, 양자역학을 이해하지 못하는 것이다"라는 말을 꺼내도록 하였다. 이런 으스스한 이상함에도 불구하고, 이론의 핵심은 몇 가지 간단한 방정식으로 축소될 수 있다. 그중 가장 유명한 슈뢰딩거 방정식은 뉴턴의 법칙이 보다 친숙한 인간 척도에서의 물체의 행동을 설명하는 것과 같은 방식으로 양자 척도에서의 입자의 행동을 설명한다. 이 방정식의 해석은 끝없이 머리를 긁을 수 있지만 수학은 훨씬 쉽게 다룰 수 있어 학생들의 철학적 질문에 눌리면 교수들이 '닥치고 계산하라'는 일반적인 권유를 하게 된다.
이와 같은 여러 방정식은 원자핵 수준에서 우리 주변에서 볼 수 있는 모든 친숙한 물질의 행동을 설명하기에 충분하다. 이들의 반직관적 성질은 특이한 현상(초전도체, 초유체, 레이저, 반도체 등)에 해당한다. 하지만 화학의 기본 구성 요소(buliding block)인 보잘것없는 공유 결합조차도 전자의 양자 상호작용의 결과이다. 1920년대에 이러한 규칙들이 마련되자, 과학자들은 처음으로 화학이 어떻게 작용하는지에 대한 상세한 이론을 갖고 있다는 것을 깨달았다. 원칙적으로, 그들은 서로 다른 분자에 대해 이 방정식을 설정하고, 시스템의 에너지를 해결하며, 어떤 분자가 안정적이고 어떤 반응이 자연적으로 일어날지 알아낼 수 있었다. 그러나 그들이 실제로 이 방정식에 대한 해답을 계산하기 위해 자리에 앉았을 때, 그들은 가장 간단한 원자(수소)에 대해 정확히 할 수 있지만 ,사실상 아무것도 없다는 것을 알게 되었다. 다른 건 다 너무 복잡하였다.
폴 디락(Paul Dirac)은 그 당시의 낙관론을 다음과 같이 요약했다.
그는 "물리학의 상당 부분과 화학의 전체 이론에 필요한 물리 법칙은 완전히 알려져 있다"며 "어려운 점은 이 법칙의 정확한 적용은 너무 복잡해서 풀 수 없다는 것이다"고 말했다. 따라서 양자역학을 적용하는 대략적인 실용적인 방법을 개발하는 것이 필요하다라는 점이다."
-폴 디락, 1929
많은 사람들이 디랙의 말을 믿었고, 곧 물리학자들은 분자 결합과 다른 화학 현상의 질적 거동에 근접할 수 있는 수학적 기법을 만들었다. 이 방법들은 입문 화학에서 친숙할 수 있는 전자가 어떻게 반응하는지에 대한 대략적인 설명에서 시작되었다. 이 설명에서, 각 전자는 특정 오비탈에 할당되며, 이는 원자핵 근처의 어느 지점에서나 단일 전자가 발견될 확률을 제공한다. 그러면 각 궤도의 모양은 다른 모든 궤도의 평균 모양(average shape)에 따라 달라진다. 이 "평균 장(mean field)"에 대한 설명은 각 전자를 하나의 궤도에만 할당하는 것으로 취급하기 때문에, 전자가 실제로 어떻게 반응하는지에 대한 매우 불완전한 그림(imcomlete picture)이다. 그럼에도 불구하고 0.5% 정도의 오차만으로 분자의 총 에너지를 추정할 수 있다.
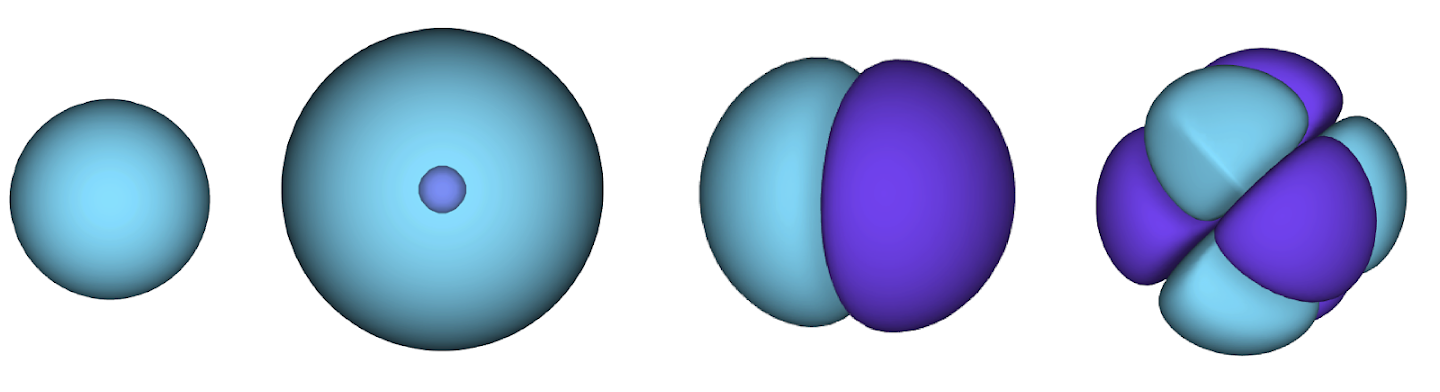
하지만 유감스럽게도 0.5%의 오차는 여전히 현업 화학자에게 유용하지 않다. 분자 결합 에너지는 시스템 전체 에너지의 극히 일부에 불과하며, 분자가 안정적인지 여부를 정확하게 예측하는 것은 종종 시스템 전체 에너지의 0.001% 또는 남은 "상관" 에너지의 약 0.2%에 의해 좌우될 수 있다. 예를 들어 부타디엔(butadiene) 분자를 구성하는 전자의 총 에너지는 몰당 100,000kcal에 가깝지만, 가능한 분자 형태 간 에너지의 차이는 몰당 1kcal에 불과하다. 이는 부타디엔의 일반적인 모양을 정확하게 예측하기 위해 축구장 너비를 밀리미터까지 재는 것과 같은 수준의 정밀도가 필요하다는 것을 의미한다.
제2차 세계 대전 이후 디지털 컴퓨팅의 등장으로, 과학자들은 전자(electrons)에 대한 까다로운 필드 설명을 넘어서는 모든 계산 방법을 개발했다. 이 방법들은 약어로 된 황당한 알파벳 표현에 담겨 있지만, 일반적으로 정확성과 효율성을 상쇄(trade-off)하는 축에 있다. 극단적으로 보면 기본적으로 정확하지만 전자의 수를 기하급수적으로 늘리는 것보다 더 나쁜 확장 방법이 있어, 가장 적은 수의 분자를 제외한 모든 경우에 있어 실용적이지 않다고 볼 수 있다. 다른 극단에는 선형으로 스케일이 조정되지만 정확하지는 않은 방법이 있다. 이러한 계산 방법은 화학의 실용화에 큰 영향을 주었다 - 1998년 노벨 화학상은 이러한 알고리즘의 창시자들에게 수여되었다.
페르미온 신경망
기존의 광범위한 계산 양자 해석 도구에도 불구하고, 효율적인 표현의 문제를 해결하기 위해서는 새로운 방법이 필요하다고 느끼고 있다. 분자역학(molecular dynamics) 같은 고전적인 화학 계산 기술은 수백만 개의 원자를 처리할 수 있는 반면, 가장 큰 규모의 양자 화학 계산(quantum chemical calculations)은 가장 대략적인 방법에도 불구하고 수만 개의 전자를 처리할 수 있을 정도에 그치기 때문이다.
고전적인 분자 시스템의 상태는 쉽게 설명할 수 있다. 각 입자의 위치와 운동량을 추적하기만 하면 된다.
하지만 양자계의 상태를 나타내는 것은 훨씬 더 어렵다. 전자 위치의 가능한 모든 구성에 확률을 할당해야 하기 때문이다. 이는 모든 전자 구성에 양수 또는 음수를 할당하는 파동 함수로 인코딩해야 하는 것을 의미하며, 파동 함수 제곱은 해당 구성에서 시스템을 찾을 확률을 제공한다. 가능한 모든 구성의 공간은 엄청난 범위에 해당한다. 각 차원을 따라 100개의 점을 가진 격자로 표현하려고 한다면, 실리콘 원자에 대한 가능한 전자 구성의 수는 우주의 원자 수보다 더 많을 것이다!
여기가 바로 우리가 생각하는 딥뉴럴넷이 도움이 될 거라고 생각한 부분이다. 지난 몇 년 동안, 신경망을 통해 복잡하고 고차원적인 확률 분포를 나타내는 데 엄청난 발전이 있었다. 이제 이러한 네트워크를 효율적이고 확장 가능하게 훈련시키는 방법을 알게 되었다. 우리는 이 네트워크들이 이미 인공지능 문제에 고차원적 함수를 적합시키는 능력을 입증했다면, 아마도 양자 파동 함수(wavefunctions)을 나타내는데 사용될 수 있을 것이라고 추측했다. 주세페 칼레오와 마티아스 트로이어(Giuseppe Carleo and Matthias Troyer)와 같은 연구원들은 어떻게 현대 딥러닝이 이상적인 양자 문제를 해결하는 데 사용될 수 있는지를 보여주었다. 우리는 딥뉴럴넷을 이용하여 화학과 응축 물질 물리학의 보다 현실적인 문제를 해결하고자 하였다. 이것은 우리의 계산에 전자를 포함시키는 것을 의미한다.
전자를 다룰 때 주름이 딱 하나 있다. 전자는 파울리 배타 원리를 따라야 하는데, 이것은 동시에 같은 공간에 있을 수 없다는 것을 의미한다. 이는 전자가 페르미온으로 알려진 입자의 일종으로 양성자, 중성자, 쿼크, 중성미자 등 대부분의 물질의 구성 요소를 포함하고 있기 때문이다. 파동 함수는 비대칭이어야 한다. 두 전자의 위치를 바꾸면, 파동 함수에 -1을 곱한다. 즉, 두 전자가 서로 동시에 위(top)에 있으면, 파동 함수(및 해당 구성의 확률)은 0이 된다.
이것은 우리가 페르미온 신경망(Fermionic Neural Network) 또는 페르미넷(FermiNet)이라는 새로운 유형의 신경망을 개발해야 한다는 것을 의미한다. 대부분의 양자화학 방법에서, 반대칭(antisymmetry)은 행렬식(determinant)으로 불리는 함수를 사용하여 도입한다. 행렬식(determinant of a matrix)은 두 행을 교체(swap)할 경우 페르미온의 파동 함수처럼 출력에 -1을 곱하는 특성을 갖는다. 따라서 단일 전자 함수를 사용하여 시스템의 모든 전자에 대해 행렬식을 계산하고 모든 결과를 하나의 행렬로 압축할 수 있다. 그러면 해당 행렬식(determinant fo that matrix)은 적절한 비대칭 파동 함수가 된다. 이 접근법의 주요 한계는 슬레이터 결정인자(slater determinant)로 알려진 결과 함수가 매우 일반적이지 않다는 점이다. 실제 시스템의 파동 함수는 일반적으로 훨씬 더 복잡하다. 이를 개선하기 위한 일반적인 방법은 슬레이터 결정인자의 큰 선형 조합(때로는 수백만 개 이상)을 취하고 전자 쌍을 기반으로 간단한 보정을 추가하는 것이다. 그렇다고 해서 에너지를 정확히 계산하기에는 역부족일 수 있다.
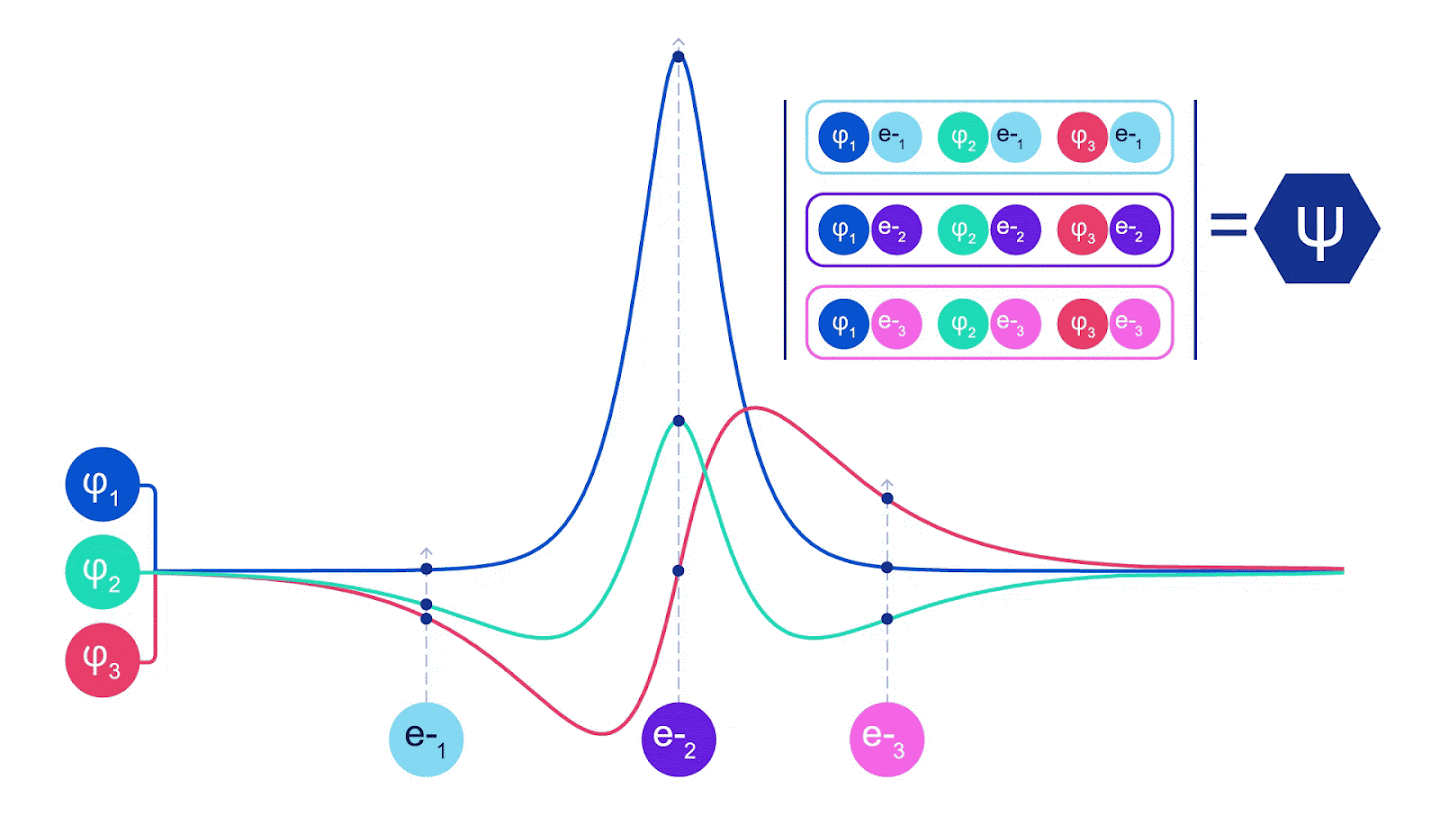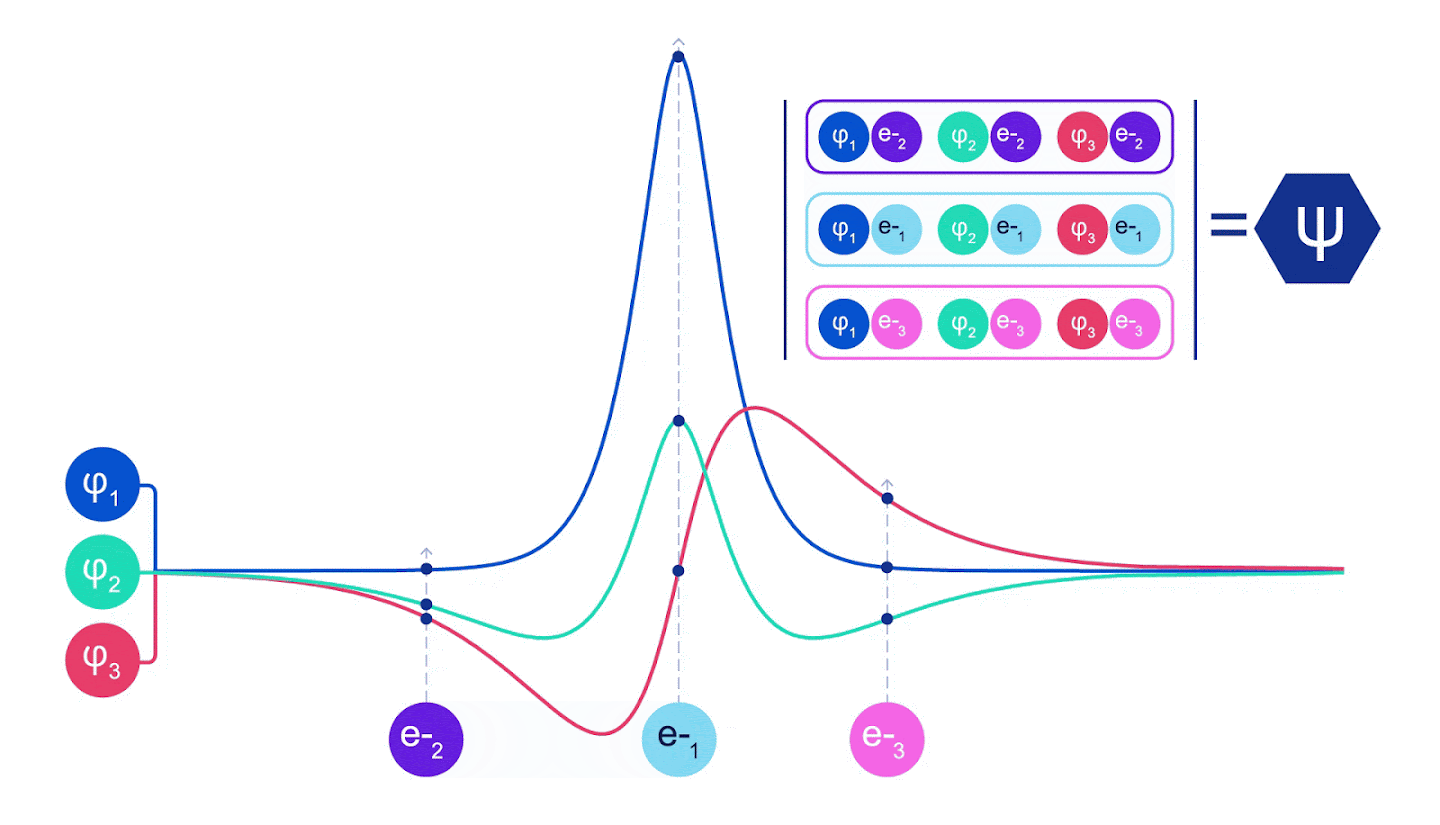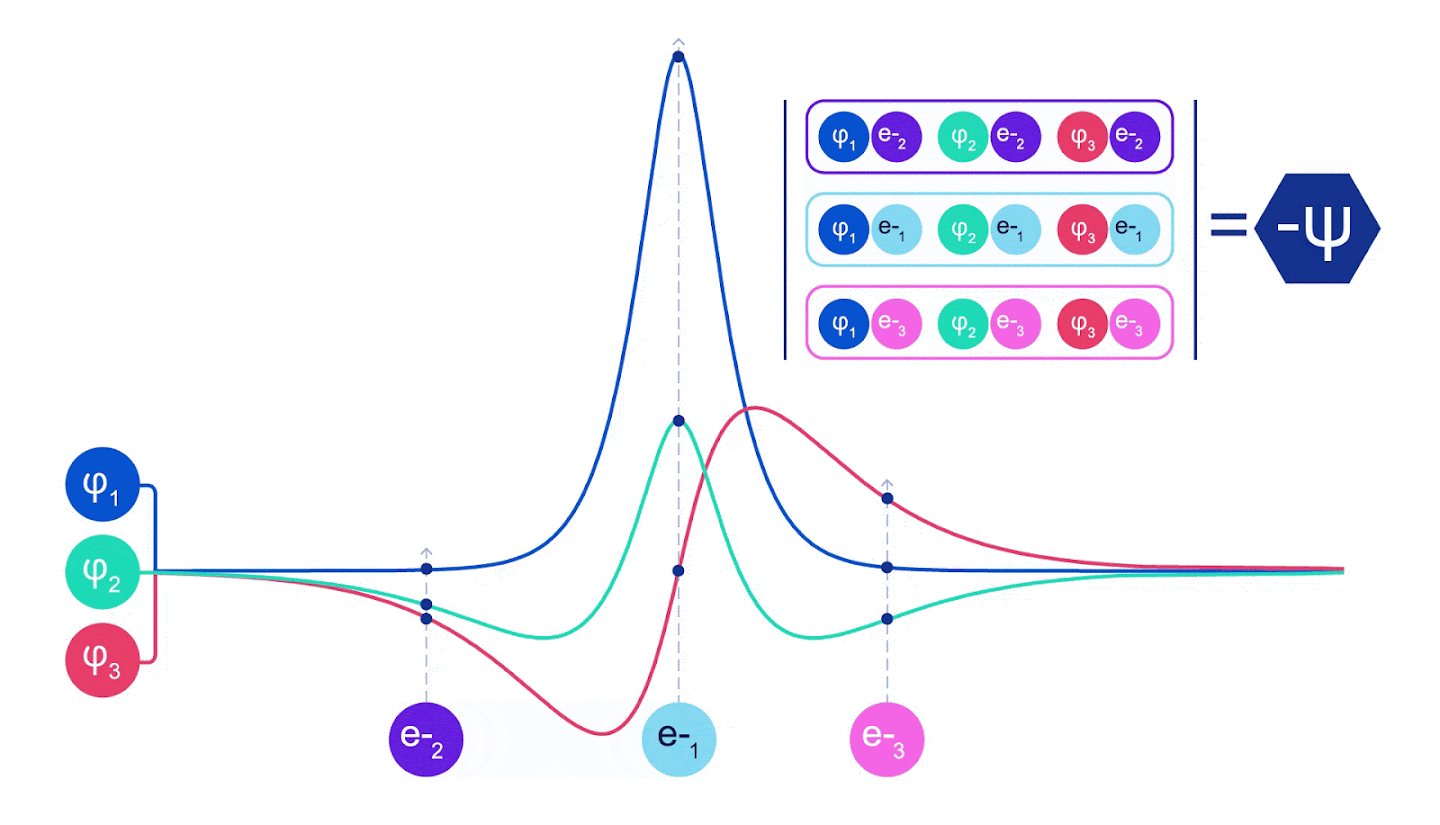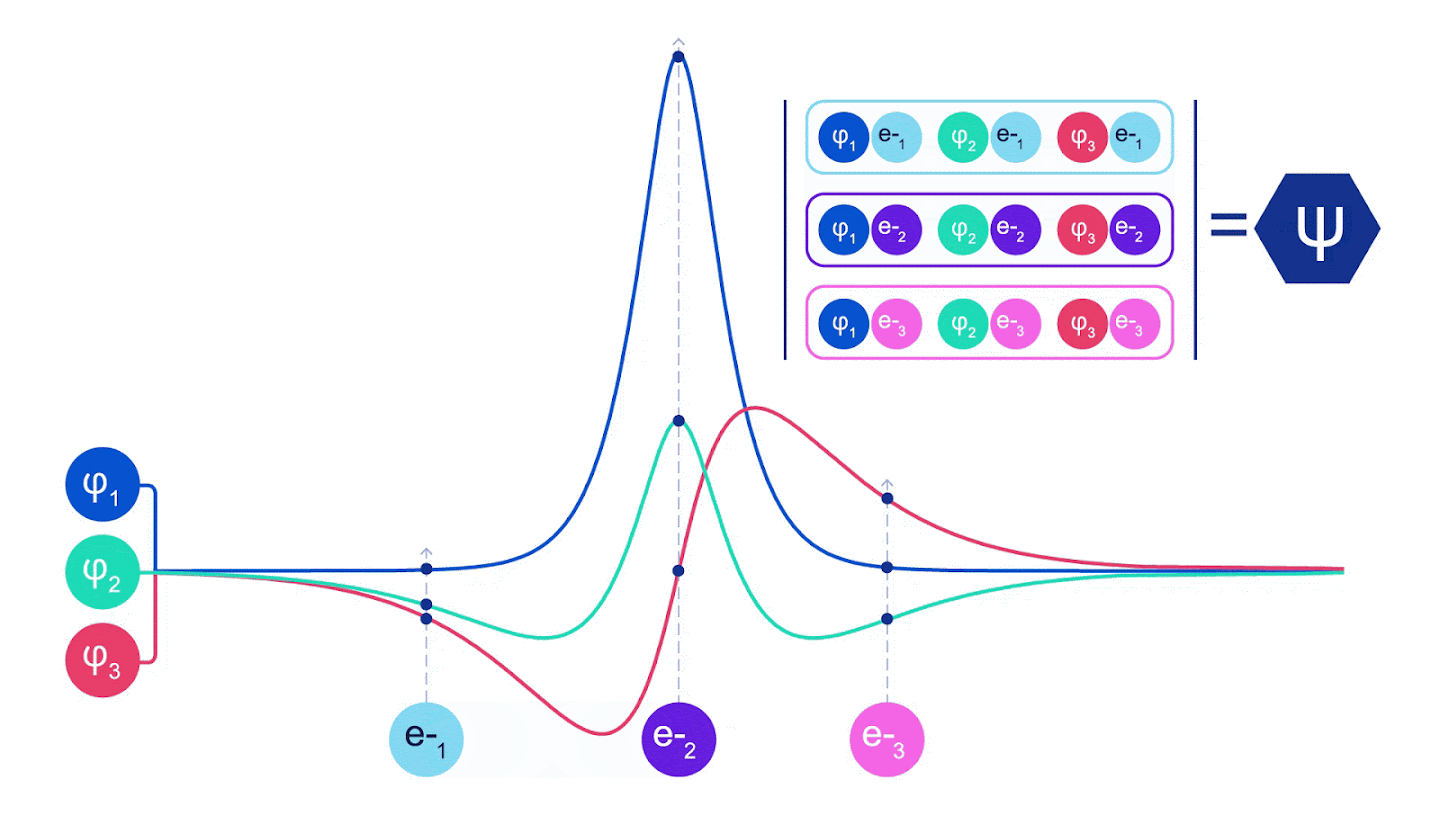
그림 2 - 슬레이터 결정체 그림 각 곡선은 그림 1의 궤도 중 하나를 통과하는 슬라이스이다. 전자 1과 2가 위치를 바꾸면 슬레이터 결정자 스왑 행과 파형 함수에 -1이 곱한다. 이것은 바울리 배척 원칙이 준수됨을 보증한다.
심층 신경망은 종종 기본 함수의 선형 결합보다 복잡한 함수를 표현하는 데 훨씬 더 효율적일 수 있다. 페르미넷에서는 각 함수를 모든 전자의 행렬식 함수(1)로 설정하여 이를 달성할 수 있다. 이는 단지 1전자 기능과 2전자 함수만을 사용하는 방식을 훨씬 뛰어넘는다. 페르미넷은 각 전자에 대해 별도의 정보 스트림을 가지고 있다. 이러한 스트림 간의 상호작용이 없다면, 네트워크는 기존의 슬레이터 결정 인자보다 더 표현력이 강하지 않을 것이다. 이 범위를 벗어나기 위해 네트워크의 각 계층에 있는 모든 스트림의 정보를 평균화하여 다음 계층의 각 스트림에 전달한다. 이렇게 하면 이러한 스트림은 비대칭 함수를 생성하기 위한 올바른 대칭 특성을 갖는다. 이것은 그래프 신경 네트워크가 각 계층에서 정보를 집계하는 방식과 유사하다. 슬레이터 결정 변수와 달리 페르미넷은 최소 신경망 층이 충분히 넓어지게 되는 한계에서 범용 함수 근사치이다. 즉, 우리가 이 네트워크를 올바르게 훈련시킬 수 있다면, 그들은 슈뢰딩거 방정식에 거의 정확한 해결책을 맞출 수 있을 것입니다.
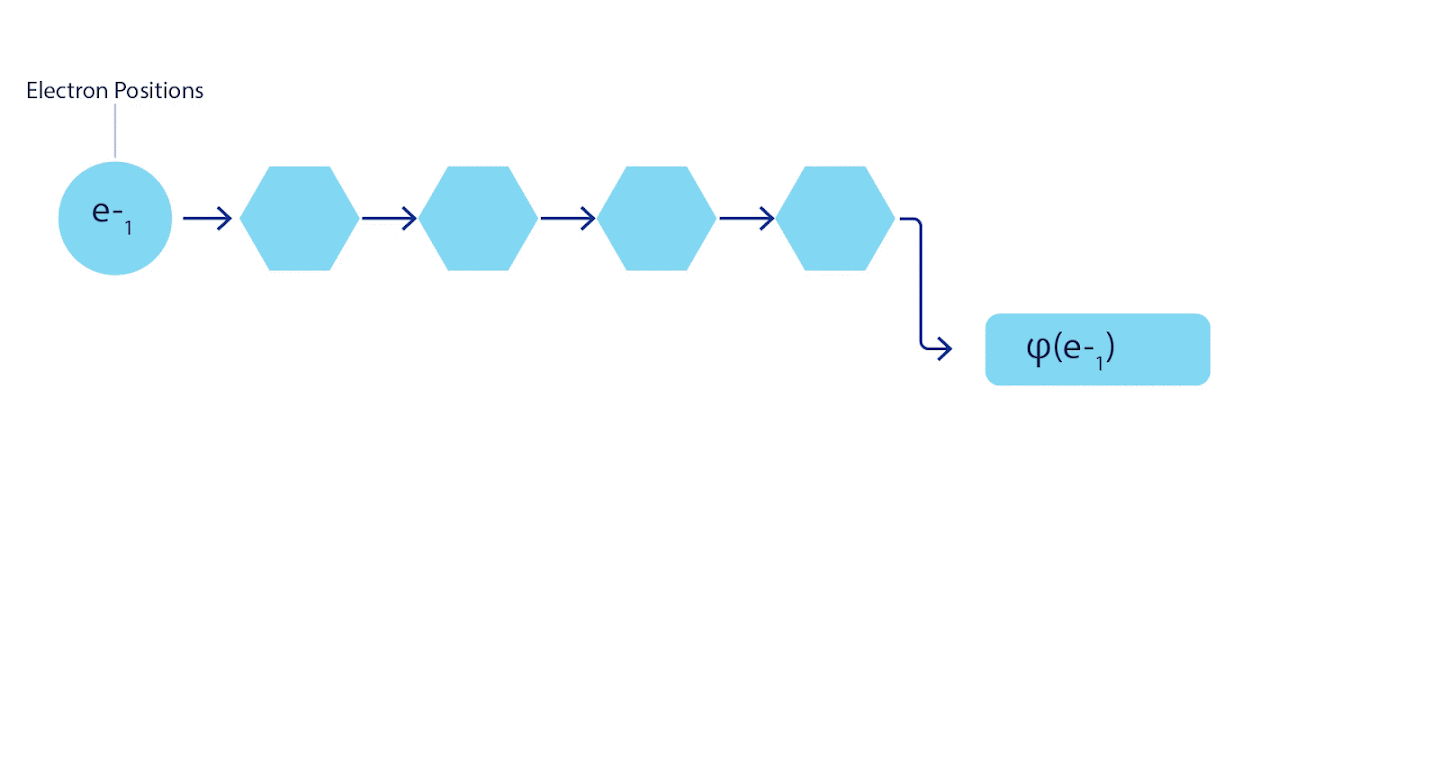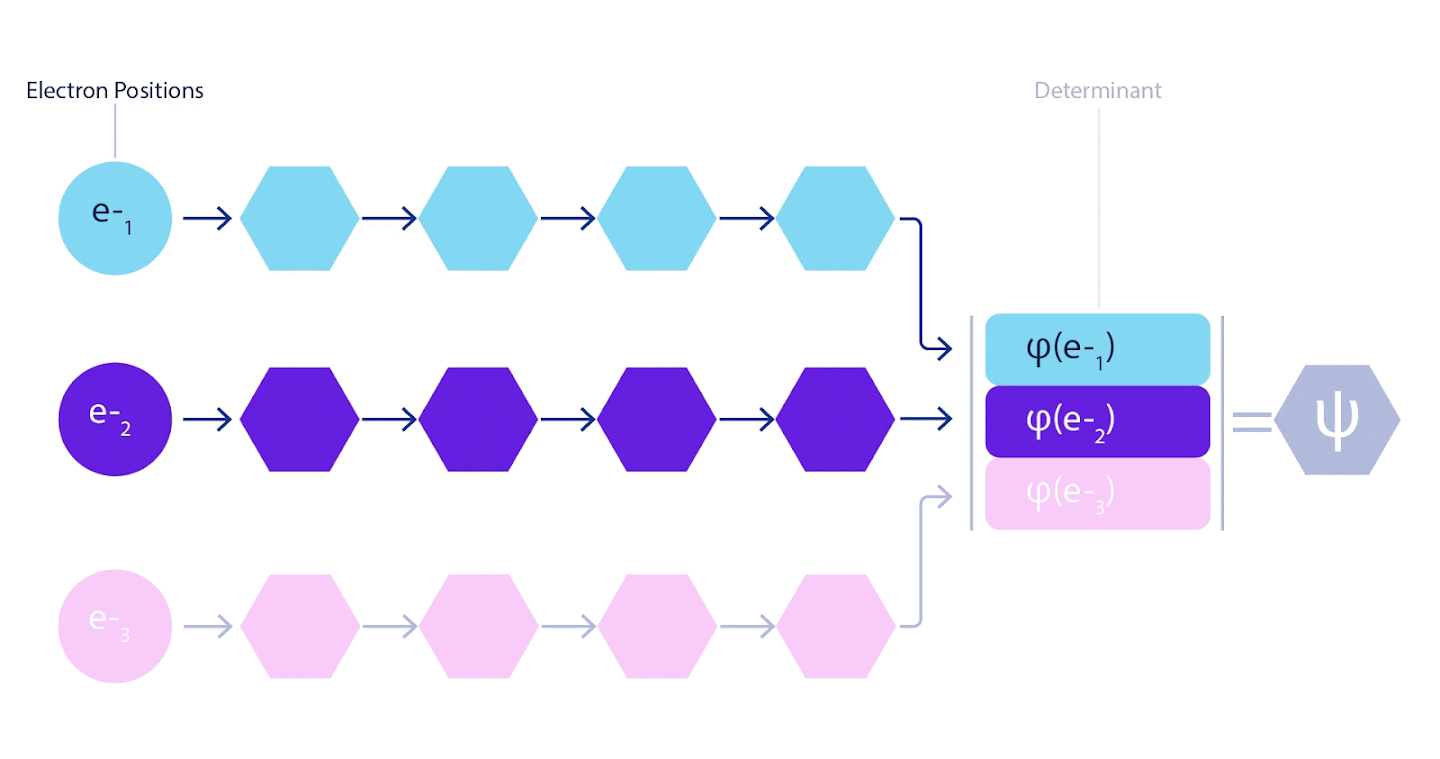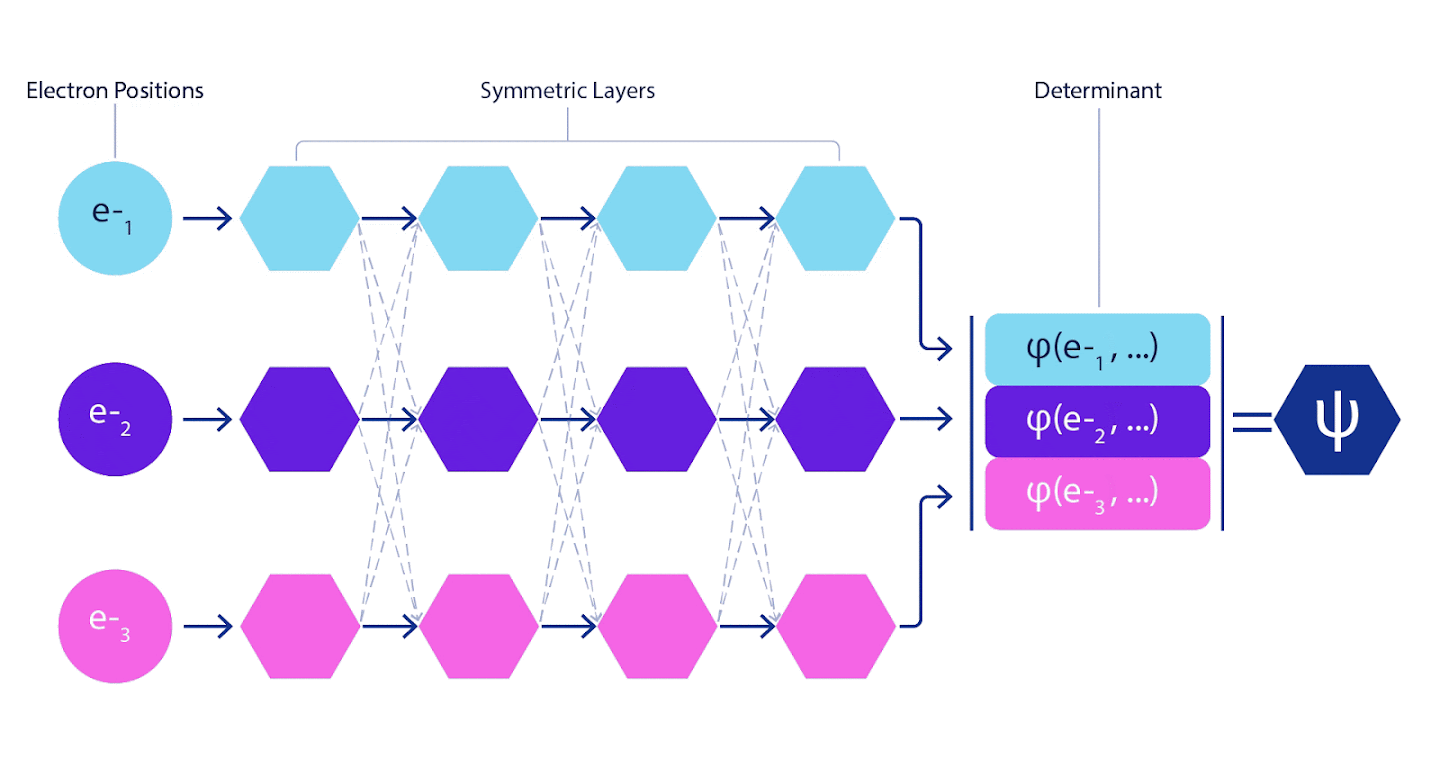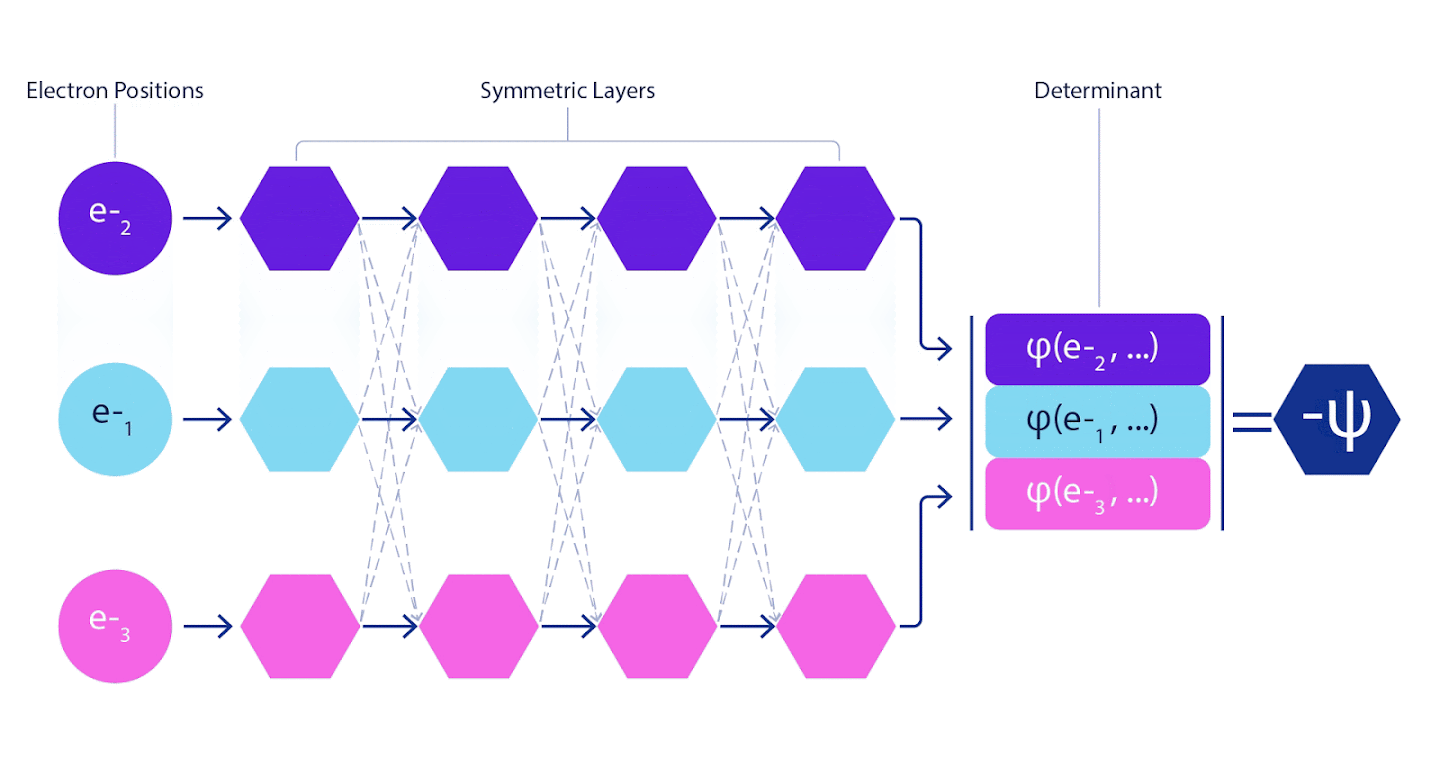
그림 3 - 페르미넷 그림
네트워크의 단일 스트림(파란색, 보라색 또는 분홍색)은 일반적인 궤도와 매우 유사하게 작동한다. 페르미넷은 스트림 사이에 대칭 상호 작용을 도입하여 파동 함수를 훨씬 더 일반적이고 표현적(expressive)으로 만든다. 기존의 슬레이터 결정자와 마찬가지로 두 전자 위치를 스와핑하면 여전히 결정자에서 두 행을 스와핑하고 전체 파동 함수에 -1을 곱한다.
우리는 시스템의 에너지를 최소화함으로써 페르미넷을 적합(fit)시킨다. 정확하게 적합시키기 위해서, 전자의 가능한 모든 구성에서 파동 함수를 계산해야 한다. 정확하지는 않더라도 대략적으로 그렇게 해야 한다.
무작위로 전자 구성을 선택하고, 각 전자 배열에서 국부적으로 에너지를 계산하며, 각 배열의 기여도를 합산하여 실제 에너지 대신 이를 최소화한다. 이것은 몬테카를로 방법으로 알려져 있는데, 도박꾼이 주사위를 계속 굴리는 것과 비슷하기 때문이다.
대략적이기는 하지만, 더 정확하게 만들어야 한다면 언제든지 다시 주사위를 굴릴 수 있다. 파동함수 제곱은 어느 위치에서나 입자의 배열을 관찰할 수 있는 확률을 제공하기 때문에 파동함수 자체에서 샘플을 생성하는 것이 가장 편리하며, 기본적으로 입자들을 관찰하는 작용을 시뮬레이션하는 것이다.
대부분의 신경망은 외부 데이터로부터 훈련되지만, 우리의 경우 신경망을 훈련시키는 데 사용되는 입력은 신경망 자체에서 생성된다. 이것은 마치 부트스트랩으로 신경망 모델을 훈련시키는 것을 의미한다. 그리고 이것은 우리가 전자들이 돌아다니는 원자핵의 위치 이외의 어떠한 훈련 데이터도 필요하지 않다는 것을 의미한다.
가변 양자 몬테 카를로(또는 줄여서 VMC)로 알려진 기본 아이디어는 60년대부터 존재해 왔으며, 일반적으로 저렴하지만 정확한 시스템 에너지 계산 방법으로 여겨지지 않는다. Slater 결정 인자에 기반한 간단한 파동 함수를 FermiNet으로 대체함으로써 지금까지 살펴본 모든 시스템에서 이 접근 방식의 정확도를 획기적으로 높였다.

그림 4 – 페르미넷(FermiNet)에서 샘플링된 시뮬레이션 전자가 바이클로부탄(bicyclobutane) 분자 주위에서 움직이고 있다.
FermiNet이 실제로 최첨단 기술을 나타내는지 확인하기 위해, 우리는 주기율표의 첫 번째 행에 있는 원자처럼 단순하고 잘 연구된 시스템을 대상으로 조사하는 것으로 시작했다. 이러한 시스템은 전자 10개 이하의 작은 시스템으로, 가장 정확한(그러나 지수 스케일링) 방법으로 처리할 수 있을 정도로 간단한다. FermiNet은 비교 가능한 VMC 계산을 큰 차이로 능가하며, 지수 스케일링 계산에 상대적인 오차를 절반 이상 줄이는 경우가 많다. 대규모 시스템에서는 지수 확장 방법이 다루기 어려워지므로 대신 "커플링 클러스터" 방법을 기준으로 사용한다. 이 방법은 안정된 구성의 분자에서는 잘 작동하지만, 결합(bonds)이 늘어나거나 끊어질 때 어려움을 겪기 때문에 화학 반응을 이해하는 데 매우 중요하다. 기하급수적인 것보다 훨씬 잘 스케일링되지만, 우리가 사용한 특정한 커플링 클러스터 방법은 여전히 7차 전력으로 상승하는 전자 수에 따라 스케일링되므로 중간 크기의 분자에만 사용할 수 있다. 우리는 수소화 리튬을 시작으로 30개의 전자를 가진 가장 큰 시스템인 바이클로부탄에 이르기까지 점차적으로 더 큰 분자에 페르미넷을 적용했다. FermiNet은 가장 작은 분자에 대해 결합된 클러스터 에너지와 단일 슬레이터 결정 요소에서 얻은 에너지 간 놀라운 99.8%의 차이를 포착했다. 바이클로부탄에서 페르미넷은 여전히 97% 이상의 상관 관계 에너지를 포착했다. 이는 아마도 "연산이 상대적으로 적지만 부정확한" 접근 방식으로는 엄청난 성과이다.
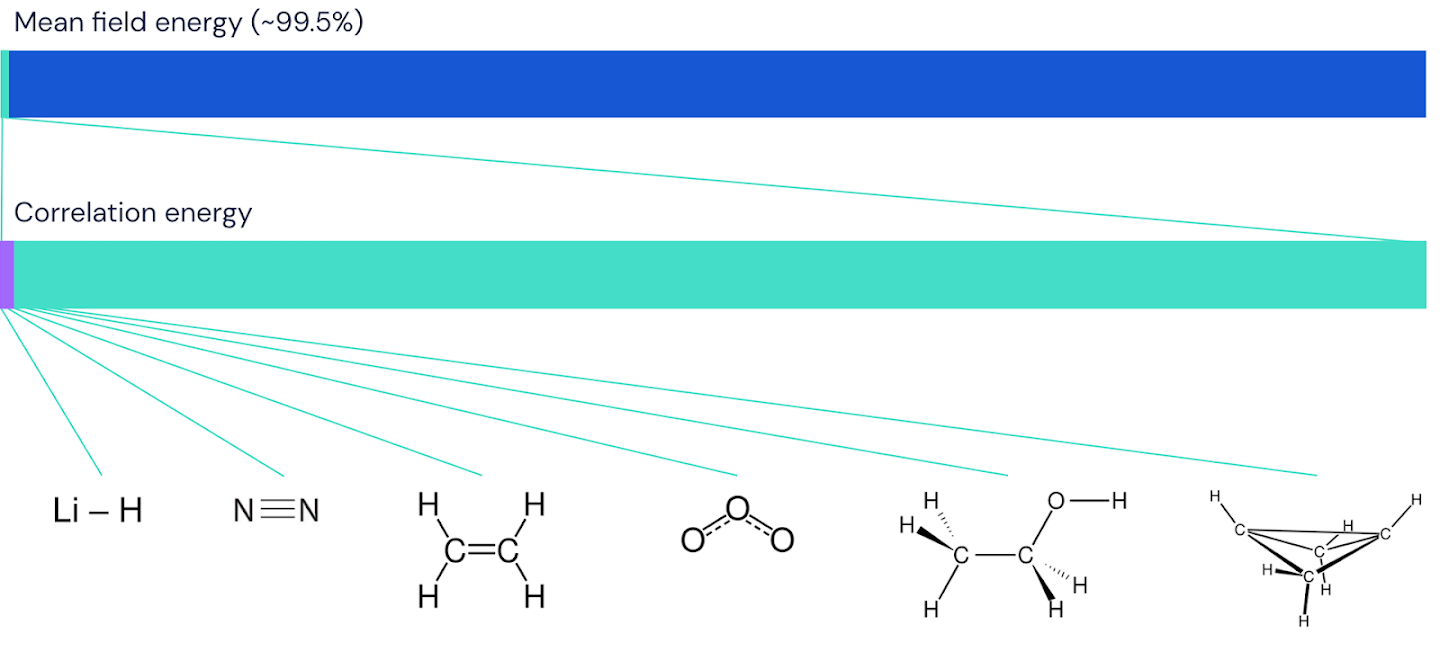
그림 5 – 페르미넷(FermiNet)이 분자에 대해 포착한 상관 에너지 분율을 그래픽으로 표현한 결과이다. 보라색 막대는 상관 관계 에너지의 99%를 나타낸다.
왼쪽에서 오른쪽으로: 리튬 하이드라이드, 질소, 에틸렌, 오존, 에탄올 및 바이클로부탄.
결합 클러스터 방법은 안정적인 분자에 잘 작동하지만, 계산 화학의 진정한 관심사항은 분자가 어떻게 늘어나거나 비틀리고 부서지는지를 이해하는 것이다. 결합된 클러스터 방법은 종종 어렵기 때문에 가능한 많은 기준과 비교하여 일관된 답을 얻어야 한다. 두 가지 벤치마크 스트레치 시스템, 즉 질소 분자(N2)와 10개의 원자가 있는 수소 체인(H10)을 살펴보았다. 질소 원자는 각각 3개의 전자를 기여하기 때문에 질소는 특히 어려운 분자 결합이다. 한편 수소 체인은 전자가 물질에서 어떻게 반응하는지 이해하는 데 관심이 있으며, 예를 들어 물질의 전기 전도 여부를 예측한다고 하자. 두 시스템 모두에서 결합된 클러스터는 평형 상태에서는 잘 작동했지만 결합이 늘어나면서 문제가 발생했다. 기존 VMC 계산은 전반적으로 좋지 않았다. 그러나 페르미넷은 결합 길이와 상관없이 조사된 가장 좋은 성능을 보여준 모델링 방법 중 하나였다.
결론
페르미넷(FermiNet)은 딥러닝과 계산 양자 화학의 융합을 위한 위대한 것의 시작이라고 생각한다. 지금까지 살펴본 대부분의 시스템은 잘 연구되고 잘 이해가 되었다. 그러나 다른 분야에서 깊이 있는 학습을 통해 얻은 첫 번째 좋은 결과가 후속 작업의 폭발적 증가와 빠른 진전으로 이어졌듯이, 우리는 페르미넷이 스케일업과 새롭고 더 나은 네트워크 아키텍처에 대한 많은 아이디어에 영감을 줄 수 있기를 희망한다. 이미 지난해 arXiv에 처음 작업을 시작한 이후 다른 그룹들은 다전자 문제에 대한 1차 원리 계산에 딥러닝을 적용하는 접근 방식을 공유하였다. 우리는 또한 계산 양자 물리학의 표면을 긁어냈으며, 페르미넷을 물질 과학과 응축 물질 물리학의 어려운 문제에도 적용할 수 있기를 기대한다. 대부분 실험에 사용된 소스 코드를 공개함으로써 다른 연구자들이 우리의 작업을 기반으로 하고 우리가 꿈도 꾸지 못했던 새로운 응용 프로그램을 시도할 수 있기를 바란다.
여기서 신문을 읽고 코드를 보십시오. 짐 킨빈, 아담 케인, 도미닉 발로우의 수치 덕분입니다.
각주
(1) 페르미넷은 또한 모든 전자 쌍에 대해 스트림을 가지고 있으며, 이러한 스트림의 정보는 다시 단일 전자 스트림으로 전달된다. 단순성을 위해 블로그 게시물에서 이를 시각화하지 않기로 선택했지만 자세한 내용은 문서에서 확인할 수 있다.

댓글 없음:
댓글 쓰기